Prof. Manju Bansal Lab, MBU, IISc, Bangalore, India.


![]()
![]()
![]()
![]()
![]()
![]()
![]()
|
Prof. Manju Bansal Lab, MBU, IISc, Bangalore, India.
|
|
|
About research area of the group
DNA structures from A to Z The right-handed double-helical Watson-Crick model for B-form DNA is the most commonly known DNA structure. In addition to this classic structure, several other forms of DNA have been observed and it is clear that the DNA molecule can assume different structures depending on the base sequence and environment. The various forms of DNA have been identified as A, B, C etc. In fact, a detailed inspection of the literature reveals that only the letters F, Q, U, Vand Yare now available to describe any new DNA structure that may appear in the future. It is also apparent that it may be more relevant to talk about the A, B or C type dinucleotide steps, since several recent structures show mixtures of various different geometries and a careful analysis is essential before identifying it as a `new structure'. This review provides a glossary of currently identified DNA structures and is quite timely as it outlines the present understanding of DNA structure exactly 50 years after the original discovery of DNA structure by Watson and Crick. Our lab is involved in modelling and simulation of various unusual DNA structures.
------------------------------------------------------------------------------------------------------------------
------------------------------------------------------------------------------------------------------------------ Structural properties of promoters: similarities and differences between prokaryotes and eukaryotes During the process of transcription, RNA polymerase can exactly locate a promoter sequence in the complex maze of a genome. Several experimental studies and computational analyses have shown that the promoter sequences apparently possess some special properties like unusual DNA structures and low stability, which make them distinct from the rest of the genome. But most of these studies have been carried out on a particular set of promoter sequences or on promoter sequences from similar organisms. To examine whether the promoters from a wide variety of organisms share these special properties, we have carried out an analysis of sets of promoters from bacteria, vertebrates and plants. These promoters were analyzed with respect to prediction of three different properties viz. DNA curvature, bendability and stability, which are relevant to transcription. All the promoter sequences are predicted to share certain common features like stability and bendability profiles, but there are significant differences in DNA curvature profiles and nucleotide composition between the different organisms. These similarities and differences are correlated with some of the known facts about transcription process in the promoters from the three groups of organisms.
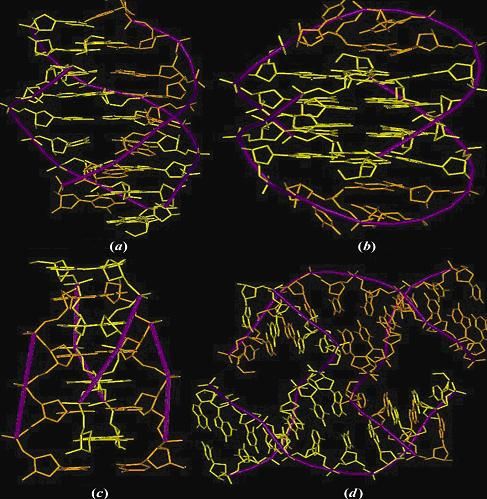
The DNA structures generated using the in-house software �NUCGEN� (Bansal et al. 1995) for: (a) an almost straight 96-mer with repetitive sequence d(CTTTTAAAAG)n (b) a 96-mer with the repetitive sequence d(CAAAATTTTG)n showing large curvature (Hagerman 1986) and (c) a 211 base pair long kinetoplast DNA fragment (Linial and Shlomai 1988). In case of all three structures only the C1' atoms are shown and the 5' end is indicated Thus though the overall function seems to be conserved across the different groups of promoters, they do differ in finer details. In eukaryotes, the DNA is packed into nucleosomes, which blocks the recognition of the core promoters by the basic transcription machinery. In comparison, the prokaryotic DNA is essentially naked, hence the RNA polymerase is not greatly hindered in its ability to gain access to the DNA and initiate RNA synthesis (65). DNA flexibility is also known to play a role in nucleosome formation and perhaps overall higher flexibility of downstream regions in eukaryotic promoters (matching that of shuffled DNA) is important in this regard. In contrast, the prokaryotic promoters, where the DNA is not packaged into nucleosomes, are overall more rigid than the shuffled sequences. Another noticeable feature of eukaryotic promoters is the presence of regulatory sites hundreds of base pairs upstream from TSS, while the regulatory elements in bacterial promoters tend to be located in the vicinity of the TSS. Our analysis also indicates that the special upstream features seem to extend at least up to -500 position in case of eukaryotic promoter sequences but seem to be confined up to -300 position in case of prokaryotic promoters. The observation that in eukaryotes, transcription factors can bind hundreds of base pair upstream seems to be reflected in the position of predicted curved region. Both groups of eukaryotic promoters show presence of a curved region considerably upstream of the TSS (> -200bp), on the other hand, the bacterial promoters show presence of a curved region nearer to the TSS.
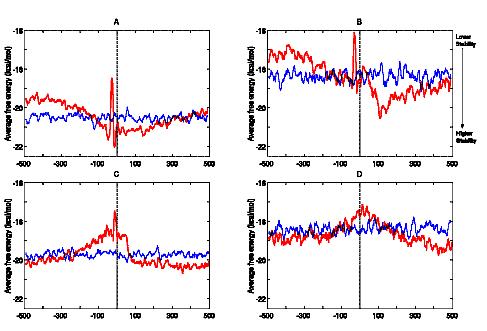
Distribution of free energy around transcription start site calculated using unified parameters. The figure shows the average free energy profiles (red) with respect to the relative base position (x-axis), in case of A) Vertebrate B) Plant C) Escherichia coli and D) Bacillus subtilis promoters. More negative values indicate greater stability (indicated by black arrow on the top right hand corner of the figure). The profiles calculated for the shuffled sequences are also shown (blue) in each case. (A. Kanhere and M. Bansal (2005) Nucleic Acids Res. 33, 3165-3175)
The observed differences between the promoters from the two bacterial origins may be attributed to the differences in their mode of binding to their respective RNA polymerases. On the other hand differences between the two eukaryotic promoter sequences viz., vertebrate and plant, may be a consequence of basic differences in their transcription regulation mechanisms, as well as due to their distinct composition (with plants being overall AT-rich and more so in the upstream region). The recent releases of plant genome sequences have revealed that plants have much higher gene density when compared to animal genomes However the increase in gene density comes at the cost of greater logistic problem in transcriptional regulation of the genes. One solution to this problem would be to have a larger number of regulatory proteins and plant genomes are in fact known to have a very high percentage of genes coding for transcription factors. We suggest that the sharp delineation in the various properties such as stability, bendability etc. of intergenic and coding region of plant genomes may be one more way of identifying the transcriptional regulatory regions. In line with this hypothesis it is to be noted that, the vertebrate class has an average gene density much smaller than the members of other three groups (Gene densities: Human ~1/100,000, Arabidopsis ~1/4000, Escherichia coli ~1/1000, Bacillus subtilis ~1/1000) and accordingly vertebrate promoters do not show large differences in their upstream and downstream region, as compared to the other three groups of promoters.
A Novel Method for Prokaryotic Gene Prediction Based on DNA Stability: In the post-genomic era, correct gene prediction has become one of the biggest challenges in genome annotation. Improved promoter prediction methods can be one step towards developing more reliable ab initio gene prediction methods. Our analysis of structural properties of promoter regions in genomic DNA clearly shows that the change in stability of DNA seems to provide a much better clue than usual sequence motifs, such as Pribnow box and +/- 35 sequence, for differentiating promoter region from non-promoter regions. It is more general and is likely to be applicable across organisms. Hence incorporation of such features in addition to the signature motifs can greatly improve the presently available promoter prediction programs.
Based on our analysis of structural properties of promoter regions in genomic DNA, we have devised a novel prokaryotic promoter prediction method based on DNA stability. The promoter region is less stable and hence more prone to melting as compared to other genomic regions. Our analysis shows that a method of promoter prediction based on the differences in the stability of DNA sequences in the promoter and non-promoter region works much better compared to existing prokaryotic promoter prediction programs, which are based on sequence motif searches. At present the method works optimally for genomes such as that of Escherichia coli, which have near 50 % G+C composition and also performs satisfactorily in case of other prokaryotic promoters.
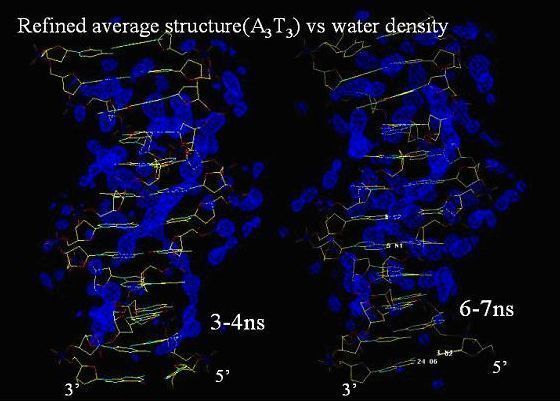
Conformational flexibility and curvature of A-tract containing DNA An understanding of the sequence dependent conformational flexibility of DNA is of great interest, since most of the biological processes like transcription, packaging, replication and repair are centered on DNA-protein interaction involving mutual structural adaptation. For more than two decades, much research has been focused on understanding B-DNA fragments containing AT base pairs because of their unique structural properties and it's implication in several biological processes. For example A-tracts are reported to play crucial role in nucleosome structure in forming complexes with regulatory proteins and as sites for minor groove binding drugs. The structure of A-tract oligonucleotide sequences has also attracted wide interest as a consequence of the linkage between A-tracts and DNA curvature. To further investigate this aspects, molecular dynamics studies were done on the sequences d(CGCAAATTTGCG) and d(CGCAAAUUUGCG) using the latest AMBER 8.0 package. The A3T3 sequence serves as the preferential binding site for several drugs which bind to AT rich minor groove regions of the DNA duplex. MD simulations were performed on A3dU3 to verify whether the exceptionally stable backbone conformations observed in steps with dT base of A3T3 were due to the presence of methyl group in Thymine. The long term aim is to study the curvature of the A-tract region for a long repetitive sequence using the structures obtained from the above simulations as starting structures. The backbone RMSD was calculated with respect to the minimized structure, excluding the terminal basepairs, for all the three simulations. A3T3 (Berendsen) has a fairly steady RMSD with a mean value of 2.5 +/- 0.4Å. A3T3 (Langevin) has a mean RMSD of 2.4 +/- 0.5Å. A3dU3 (Langevin) has a mean RMSD of 2.6 +/- 0.6Å. The higher mean and standard deviation in the last case is due to very high fluctuations in the terminal base-pairs which also affects the second base-pairs on either side. (On including the terminal base-pairs, the RMSD values increase to 2.9 +/- 0.5Å, 3.2 +/- 0.6Å and 3.6 +/- 1.1Å respectively.) Role of dynamic fluctuations on DNA grooves and its implications for ligand binding Sequence dependent conformational flexibility of DNA is of great interest due to its implications for drug-DNA and DNA-protein interactions. An understanding of the environment dependent sensitivity of DNA structure is necessary to elucidate its structure-function relationships. To understand the dynamics, hydration and ion binding features of A-tract and non A-tract sequences, 7ns Molecular Dynamics (MD) study have been performed on three dodecamer sequences: d(CGCAAATTTGCG)2 (A3T3) and on heteropolymers containing AT and CG sequences, d(CGCATATATGCG)2 (AT)3 and d(CGCGCGCGCGCG)2 (CG)3 .
The results of the MD on the A-tract sequence suggest that the intrusion of Na+ ions into the minor groove is a rare event and the structure is not very sensitive to the location of the sodium ions. Depending on the minor groove width, specific hydration patterns are seen. "Spine of hydration" is seen near the A-rich region and "ribbon of hydration" near the alternating AT and GC regions. The characteristic narrowing of the minor groove in the A-tract region is seen to precede the formation of the spine of hydration. The C2-H2 - O2 hydrogen bonds in the minor groove of A-tract sequences is found to occur even before the narrowing of the minor groove, indicating that such interactions are an intrinsic feature of A-tract sequences. Greater fluctuations (based on standard deviations) of slide, roll and twist values are observed for the dinucleotide steps spanning the AT tracts than the A and CG tracts. The absence of cross-strand hydrogen bonds and the greater fluctuations of the base pair parameters of (AT)3 sequence shows the intrinsic flexibility of the sequence. Thus from the MD studies, it can be concluded that the (AT)3 sequence is more flexible than A3T3 and (CG)3 sequences.
All the three MD studies showed fluctuations in the backbone torsion angles, which correspond to one of the conformational substates of B-DNA, known as BII conformation. Analysis on MD and crystal structures shows that BII conformation is sequence specific and is characterized by high twist, negative roll and positive slide values. Interestingly, the magnitude of variation in the dinucleotide parameters is seen to depend mainly on two factors, the magnitude of e-z difference and the presence or absence of BII conformation across the WC base-paired dinucleotide step.These variations in the dinucleotide parameters also lead to minor groove width variation that could be implicated in ligand binding, both of proteins and drug molecules. The role of B II nucleotides in C-form of DNA has also been examined, a model for C-form DNA has been built for a dodecamer d(CGCGCGCGCGCG)2 with all the steps in BII backbone conformation. The MD study on the model showed that duplex DNA structure with BII backbone in all the steps may not be a stable model and hence the C-form should have a mixture of BI and BII backbone conformations.
Assignment of Secondary Structure in Proteins There are several methods available (viz. DSSP, STRIDE, DEFINE) to identify the protein secondary structure elements given the atomic co-ordinates of the protein molecule. However using these algorithms it is difficult to assign the N- and C- terminii of the secondary structure motifs, as well as other geometrical parameters. We have developed an extension to our earlier algorithm HELANAL in order to more precisely ASSIGN the various regular secondary structure elements in proteins, particularly helices using only the geometric path traced by C-alpha atoms. These programs are being used to study the occurrence of different type of helices in proteins as well as their distortions in globular proteins.
|
|
Copyright © 2008
Prof. Manju Bansal
|