| ABOUT
THE PROGRAM: a) Standalone version b) Web-version The NUPARM program has been developed so as to meet most of the requirements stipulated at the EMBO Workshop on DNA Curvature and Bending, held at Cambridge, U.K., in Sept. 1988. The Nomenclature and Description of the DNA structural parameters follows the Cambridge Convention ( Dickerson 1989,NAR ) 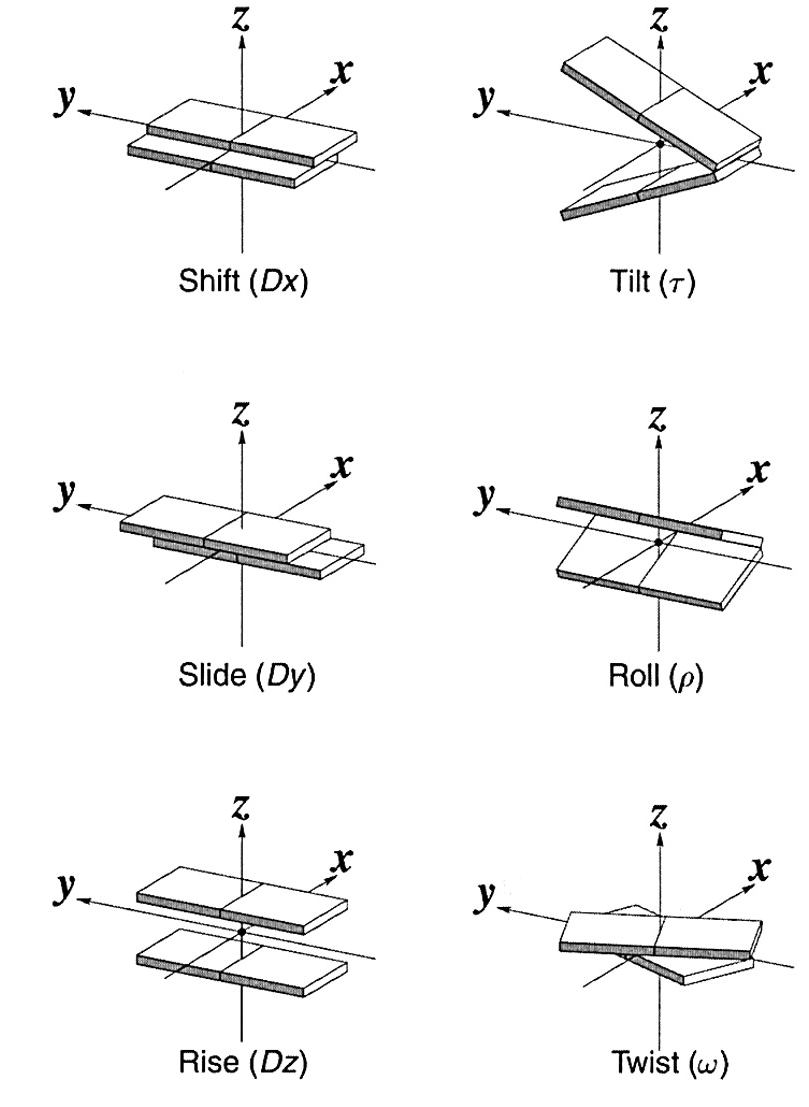 
1. Base step and base pair parameters:
Tilt, Roll, Twist, Shift, Slide and
Rise.
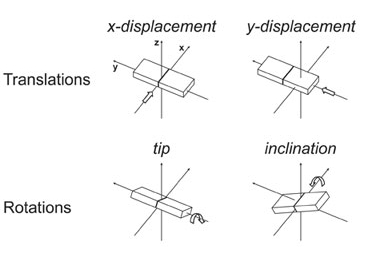
Inclination, Tip, Helical Twist,
Displacements (dx and dy) and Helical Rise (dz).
Virtual glycosidic bond angles: between the two glycosidic bonds (N--C1') and the
C1'--C1' vector are also given. The Intra-basepair C8--C6 and C1'--C1' distances are also listed
* Opening angle is defined as the angle between the lines C1'--C8 and C1'--C6 projected on to the mean base-pair plane.
Inclination, Tip, Helical Twist, Displacements (dx and dy)
and Helical Rise (dz).
2. Single-strand parameters: (for strand 1 and strand 2)
The
cylindrical polar coordinates of the phosphate and C1' atoms are calculated by assuming
the global axis to be the Z-axis,
4. Inter-chain P--P anc C1'-C1' distancesThe intra and inter-chain phosphate distances are also listed, for P and C1' atoms. 5. Backbone and Glycosidic Torsion Angles The torsion angles is used, then the main chain torsion angles, the glycosidic torsion angles and the furanose ring, endocyclic torsion angles are printed. The sugar pucker phase angle and amplitude of puckering (as described in Harvey and Prabhakaran, J. Amer. Chem. Soc. 1986) are listed along with the ring torsion angles. This calculation is the most time consuming part of the analysis program. 6. Additional information: The direction cosines of the local helix axes and base-pair normals as well as the coordinates of local helix origins and base-pair centers are printed. The angle between each local helix axis and all others, as also the angles between base-pair normals are tabulated.
Only the purine/pyrimidine ring atoms are included when calculating the base normals. We found only a nominal difference in the parameters when all the pendant atoms are included. The mean base-pair normal is taken to be the average of the two normals in the base-pair, in order to minimize the differences due to the size of the purine and pyrimidine rings. The 5'-->3' direction of strand 1 is used to define the positive Z-direction and the Y-axis is taken as pointing towards this strand. The X-axis then points towards the major groove, as suggested in the Cambridge Convention( Dickerson 1989, NAR). The base-pair center is defined as the midpoint of C6 and C8 atoms of pyrimidines and purines respectively. The long axis (Y-axis) can be along the C6---C8 direction and passes through the base-pair center. The definition of the local helix and wedge parameters are in terms of the local helix axis and the mean Z-axis respectively for the doublet involved (as described by Bhattacharyya et al J. Biomol. Struct. Dynam.,1989 ). In this description the signs of tilt, tip and buckle were opposite to the Cambridge definition. Subsequently the algorithm has been modified so as to follow the Cambridge Convention, wherein all clockwise rotations are positive when viewed down the rotation axis. INPUT TO THE PROGRAM: The program next asks whether the nucleotides in the duplex are numbered according to the standard PDB 5'-->3' residue numbering scheme, with the first nucleotide base paired to the last base. If not, the file containing a user-defined, residue numbering scheme has to be specified (eg. if an AMBER output is being used, or the file has base overhangs, bulges etc, such that it is only partially paired as in tRNA and many other structures). This option can also be used to analyze duplexes in protein-DNA complex structures, if the residue numbers are unique. The residue numbers of paired bases have to be given in (I5, 1x, I5) format for a duplex. The helix can be reoriented to give the
base-pair parameters with respect to a global axis. The realignment is
done in two steps: (i) the best line is fitted (by the method of least
squares) to a set of representative points taken either from all the
residues, or the doublet steps. The molecule is rotated such that this
line becomes parallel to the global Z-axis. (ii) The same
representative points are used to find the mean point through which the
line passes, and the molecule is translated such that this point
coincides with the origin and the best line with the Z-axis.
The options to calculate P--P and C1'--C1' cylindrical polar coordinates, the intra and inter chain distances for these atoms, all backbone and sugar ring torsion angles, as well as inter base parameters in each strand are also available. OUTPUT FILES NUPARM generates two output files
Note:
|
NUPARM
A Program for analyzing sequence dependent variations in
nucleic acid
(DNA & RNA) double helices
(DNA & RNA) double helices
You are the visitor number: 
©2012 Molecular Biophysics Unit, Division of Biological Sciences, Indian Institute of Science, Bangalore-560012, INDIA
©2012 Molecular Biophysics Unit, Division of Biological Sciences, Indian Institute of Science, Bangalore-560012, INDIA