Promoter prediction methodology
The
average free energy (E) over known promoter sequences and the difference (D) between E and the average free energy over downstream random sequences (REav) are used to search for promoters in the genomic sequences. Difference in free energy or stability of neighboring regions are calculated and compared with the assigned
cutoff (obtained from the energy difference between upstream and downstream
regions in the vicinity of known TSS), to predict promoters in genomic DNA
sequences (Rangannan V and Bansal M 2007, 2009).
Free energy
(stability) calculation
The stability of a
double stranded DNA molecule can be expressed as sum of free energy of its
constituent base paired dinucleotides.
In the present
study free energy over a long
continuous stretch of DNA sequence was calculated by dividing the sequence into
overlapping windows of 15 base pairs (or 14 dinucleotide steps). The energy
values corresponding to the 16 dinucleotide steps (10 unique dinucleotide) are
taken from the unified parameters obtained from melting studies on 108
oligonucleotides (Allawi and SantaLucia 1997; SantaLucia 1998).
|
Dinucleotide step |
Free
energy
(kcal/mol) |
|
AA |
-1.0 |
|
TT |
-1.0 |
|
AT |
-0.88 |
|
TA |
-0.58 |
|
CA |
-1.45 |
|
TG |
-1.45 |
|
AC |
-1.44 |
|
GT |
-1.44 |
|
CT |
-1.28 |
|
AG |
-1.28 |
|
GA |
-1.30 |
|
TC |
-1.30 |
|
CG |
-2.17 |
|
GC |
-2.24 |
|
GG |
-1.84 |
|
CC |
-1.84 |
Threshold
calculation
Protein coding promoter sequences of 1001 nt long were grouped together on the basis of their GC composition, at 5 %GC intervals and their average free energy was calculated. The average free energies over the promoter regions (particularly the 1001 nt long regions spanning from -500 to +500 with respect to TSSs) with similar GC composition are observed to be approximately same for the above classification.
The average free energy (E) over known promoter sequences and the difference (D) between E and the average free energy over downstream random sequence (REav) were used as cut-off values. Threshold values (E and D) have been generalized for every 5 %GC interval. These cutoff values were then applied over DNA sequences of 1001 nt length (spanning over -500 to +500 w.r.t to the
annotated TSSs) as well as whole genome sequence to predict the promoter regions.
Average free
energy profile for 429 E. coli promoters that are more than 500 nt apart
(taken from EcoCyc Database version 9.1). Blue line represents the average
stability profile for shuffled sequences. E-cutoff andD-cutoffare used as threshold values to predict promoter regions.

True
positive, false positive and false negative definition for known promoter
sequences
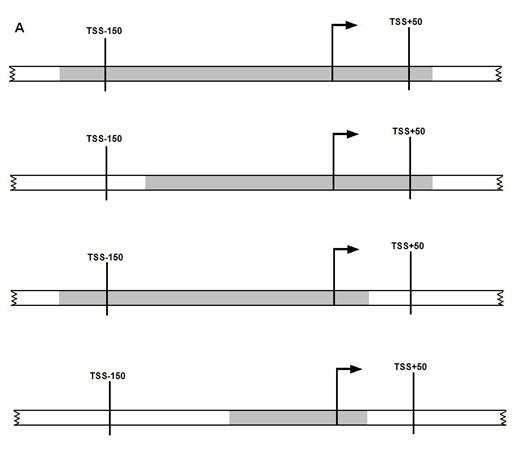
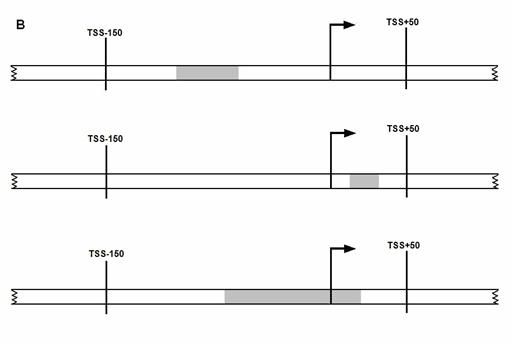
A promoter is
considered to be predicted correctly if it meets at least one of the following
three conditions (as illustrated in the figure below), (A) A transcription start
site (TSS) lies within the predicted promoter region, (B) Predicted promoter
region lies within the 200 nt region spanning from -150 nt upstream of TSS to
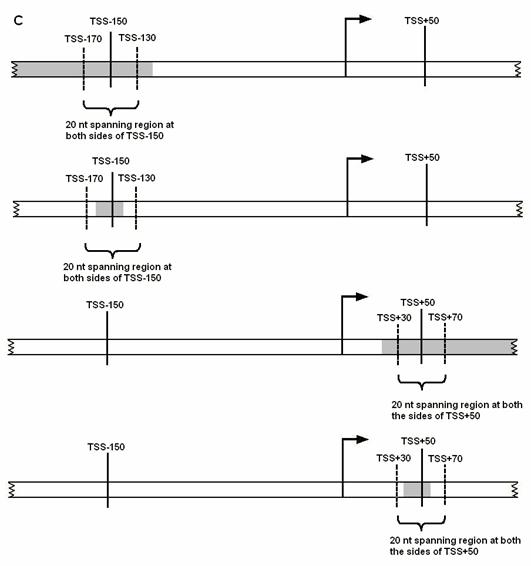
+50 nt downstream of TSS, (C) Predicted promoter region overlaps with the 200nt
region mentioned above. (i.e. at least 20 nt of predicted promoter region
overlaps with the 200 nt region, if not the overhang region outside of 200 nt
region is less than 20 nt in length). If a predicted promoter region for the
1000nt long genomic sequence (spanning the region -500 to +500 w.r.t to the
annotated TSSs) meets
any of the above criteria, then it is considered as a
true positive.
If more than one predicted region satisfies any one of
the above condition, the one nearest to TSS is considered as true positive. All
other predicted promoter signals are considered as
false positives. If no promoter signal is located in a 1000 nt long
sequence, it is considered as a case of
false negative.
Illustration
of various cases of true positive prediction. In the
following figures gray shaded region
represents the promoter region as predicted by our method. A promoter is
considered to be predicted correctly (TP) if it meets one of three conditions.